PASOA
Start of topic | Skip to actions
Provenance-Aware Bioinformatics Application
Experiment
The experiment, described below, was designed by Klaus-Peter Zauner and Stefan Artmann.Biology
Proteins are the essential functional components of all known forms of life; they are linear chains of typically a few hundred building blocks taken since 2 milliards years from the same set of about 20 different amino acids. Protein sequences are assembled following a code sequence represented by another polymer (mature mRNA). This polymer is produced by splicing certain pieces (the exons) of a molecular copy of the coding region of a gene on the DNA, while discarding other pieces (the introns) of the copy. During and following the assembly, the protein will curl up under the electrostatic interaction of its thousands of atoms into a defined but agile shape of typically 58 nm size. The resulting 3D-shape of the protein determines its function. The structure of protein sequences is of considerable interest for predicting which sections of the DNA encode for proteins and for predicting and designing the 3D-shape of proteins. For comparative studies of the structure present in an amino acid, it is useful to determine their compressibility. Compression exploits context-dependent correlations within the sequence. The fraction of its original length to which a sequence can be loss-lessly compressed is an indication of the structure present in the sequence. In general, no practical compression method can discover all structures, so actual compression of a sequence can only yield a lower bound of the sequences compressibility. For the same reason, the compressibility values are also relative to the applied compression method [9]. Methods that are good in discovering structure are computationally expensive; initial investigations on protein compressibility indicated that it is indeed difficult to discover structure in protein sequences [13]; however, recently, progress has been made by grouping amino acids [14]: if the compression of the sequences serves only to quantify structure and decompression is not intended, the sequences can be recoded with a reduced alphabet. In an amino acid sequence, for instance, each amino acid symbol is replaced by a symbol representing a group of amino acids. Compression is then applied to the recoded sequence. The results of this experiment can, for example, be used to determine the amino acid groupings that maximise compressibility.Workflow
To focus the discussion, we consider the main workflow of the comparative sequence compressibility experiment, shown in the workflow below.
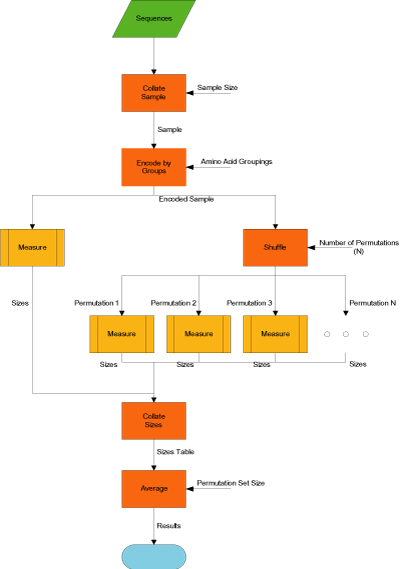
- gzip/ppmz Compression: The input sample is compressed using the gzip or ppmz algorithm.
- Measure Size: The sample and its two compressed forms are measured to determine their respective sizes.
- Collate Sizes: The size data output from all Measure Size steps are collated into a single table.
Use Cases
In this section, we introduce some use cases that we have identified for the Protein Compressibility Experiment. For a more complete survey of provenance use cases and technical requirements, we refer the reader to [11].Use Case 1
A bioinformatician, B, downloads sequence data of microbial proteins from the database RefSeq and runs the compressibility experiment. B later performs the Sample same experiment on the same sequence data, again downloaded from RefSeq. B compares the two experiment results and notices a difference. B determines whether the difference was caused by the algorithms used to process the sequence data having been changed. For instance, with Use Case 1, B could observe that the compression algorithms used in the compressibility work- flow were configured differently in two runs of the experiment, and so produced differing results.Use Case 2
A bioinformatician, B, performs an experiment on a FASTA sequence encoding a protein. A reviewer, R, later determines whether or not the sequence was in fact processed by a service that meaningfully processes protein sequences only. The principle of amino acid group encoding also applies to other types of sequences; for instance, in a nucleotide sequence, each codon triplet can replaced with a symbol representing a group of codons. A fundamental part of the experiment presented here is for symbols for the amino acids making up a protein sequence to be replaced by symbols for the groups to which they belong (i.e., the Encode by Groups activity). If a nucleotide sequence was accidentally used at this stage rather than an amino acid sequence, there would be no error in running the workflow because the symbols used for nucleotides are a subset of those used for amino acids. However, the experiment results would not be meaningful, because the groups used and workflow results compared against would all be particular to amino acids. In this case, we can say that, while the workflow is syntactically correct, it is semantically incorrect, which is what Use Case 2 aims to discover.Implementation
Grid Computing
This workflow offers great potential for massive parallelism given the vast number of input sequences and the large number of permutations to be considered. Thus, we have decided to adopt VDT (the Virtual Data Toolkit, www.cs.wisc.edu/vdt), which offers good possibility of scheduling over the Grid through the use of Condor. However, very quickly, our application has turned out to be a heterogeneous system, involving multiple technologies to run and compose applications (or services), as we now explain. While compression methods such as gzip or ppmz can run directly from the command line, other advanced compression methods may be available as Web Services. Likewise, sequences provided as inputs are typically obtained by some form of remote procedure call to a remote database. Given that for a typical sample, compression takes of the order of 100ms, we have partitioned the processing of permutations into scripts that provided a sufficient granularity of computation (the order of 15 minutes) in order to offset the overhead of grid scheduling and file transfer (cf. the dashed box in the main workflow). Consequently, this specific application relies of a variety of methods to run and compose computations: binary executables, shell scripts, Web Services and VDT/Dagman workflows.Principles of Provenance Recording
Given that multiple parts of our application can run under the control of different runtime systems, at separate locations, some mechanism is required for all these parts to contribute provenance data, so that it can be used seamlessly by programs that support use cases. However, no system supports such open-ness. For instance, while Chimera [5] provides for domain-independant provenance recording for VDL scripts, it offers no mechanism for submitting provenance information, which would have not been produced under the control of VDT. Additionally, while Chimera provides for provenance recording of activities they regard as atomic, finer grained recording may be required to follow the actual scientific process being executed. Arbitrary pieces of data (such as scripts themselves) may have to be submitted to support our use cases. Chimera records information that is useful for debugging (such as invoked commands, inputs and outputs), but does not allow arbitrary data to be submitted; alternatively, Chimeras virtual data catalog could be used to store arbitrary metadata stored, but it offers no generic mechanism for submitting metadata about a specific command execution. Finally, the different activities in a workflow should typically be grouped in different ways, with each grouping providing a well understood semantics. For instance, a workflow run is usually referred to as a session, while a sequential succession of activities as a thread. Such groupings are essential to analyse dependencies of activities while reasoning over provenance.Provenance Recording in PReServ
We used a deployment of the PReServ Web Service implementation to record provenance data in the experiment.Bibliography
- P. Buneman, S. Khanna, K.Tajima, and W. Tan. Archiving scientific data. In Proc. of the 2002 ACM SIGMOD International Conference on Management of Data, pages 112. ACM Press, 2002.
- Y. Cui, J. Widom, and J. L. Wiener. Tracing the lineage of view data in a warehousing environment. ACM Trans. Database Syst., 25(2):179227, 2000.
- R. Figueiredo, P. Dinda, and J. Fortes. A case for grid computing on virtual machines. In Proceedings of the 23rd Internatinal Conference on Distributed Computing Systems (ICDCS 2003), 2003.
- I. Foster and C. Kesselman, editors. The Grid: Blueprint for a New Computing Infrastructure. Morgan Kaufman Publishers, 1998.
- I. Foster, J. Voeckler, M. Wilde, and Y. Zhao. Chimera: A virtual data system for representing, querying and automating data derivation. In Proceedings of the 14th Conference on Scientific and Statistical Database Management, Edinburgh, Scotland, July 2002.
- M. Greenwood, C. Goble, R. Stevens, J. Zhao, M. Addis, D. Marvin, L. Moreau, and T. Oinn. Provenance of e-science experiments - experience from bioinformatics. In Proceedings of the UK OST e-Science second All Hands Meeting 2003 (AHM03), pages 223226, Nottingham, UK, Sept. 2003.
- P. Groth, M. Luck, and L. Moreau. A protocol for recording provenance in service-oriented grids. In Proceedings of the 8th International Conference on Principles of Distributed Systems (OPODIS04), Grenoble, France, Dec. 2004.
- K. Keahey, K. Doering, and I. Foster. From sandbox to playground: Dynamic virtual environments in the grid. In Proceedings of the 5th International Workshop in Grid Computing (Grid 2004),, Pittsburgh, PA, Nov. 2004.
- K. Lanctot, M. Li, and E. h. Yang. Estimating dna sequence entropy. In Proceedings of the Eleventh Annual ACMSIAM Symposium on Discrete Algorithms, pages 409418, San Francisco, California, Jan. 911, 2000. ACM.
- D. Lanter. Design of a lineage-based meta-data base for gis. Cartography and Geographic Information Systems, 18(4):255261, 1991.
- S. Miles, P. Groth, M. Branco, and L. Moreau. The requirements of recording and using provenance in e-science experiments. Technical report, University of Southampton, 2005.
- S. Miles, J. Papay, T. Payne, M. Luck, and L. Moreau. Towards a protocol for the attachment of metadata to service descriptions and its use in semantic discovery. Scientific Programming, page 14, 2005.
- C. Nevill-Manning and I. Witten. Protein is incompressible. In J. Storer and M. Cohn, editors, Proc. Data Compression Conference, pages 257266, Los Alamitos, CA, 1999. IEEE Press.
- G. Sampath. A block coding method that leads to significantly lower entropy values for the proteins and coding sections of haemophilus influenzae. In Proceedings of the Computational Systems Bioinformatics (CSB03). IEEE Computer Society, 2003.
to top
Edit | Attach image or document | Printable version | Raw text | More topic actions
Revisions: | r1.7 | > | r1.6 | > | r1.5 | Total page history | Backlinks
Revisions: | r1.7 | > | r1.6 | > | r1.5 | Total page history | Backlinks